1 afectado/a de cada 4 a 8 millones de recién nacidos
 El síndrome de progeria Hutchinson- Gilford fue descrito por primera vez en 1886 por el Dr. Jonathan Hutchinson, su nombre deriva del planteamiento propuesto en 1904 por el Dr. Hastings Gilford.
El síndrome de progeria Hutchinson- Gilford fue descrito por primera vez en 1886 por el Dr. Jonathan Hutchinson, su nombre deriva del planteamiento propuesto en 1904 por el Dr. Hastings Gilford.
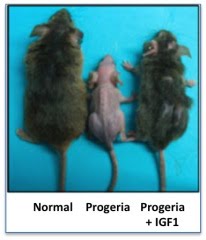
Se trata de una enfermedad genética rara caracterizada por el envejecimiento prematuro en edades tempranas. El proceso de envejecimiento en estos individuos es entre 5 y 10 veces más acelerado de lo normal.
 El síndrome de Progeria Hutchinson-Gilford debe diferenciarse del síndrome Wiedemann-Rautenstrauch conocido como síndrome neonatal progeroide, en el cual el inicio del envejecimiento comienza en el útero, y los signos y síntomas son evidentes al nacer.
El síndrome de Progeria Hutchinson-Gilford debe diferenciarse del síndrome Wiedemann-Rautenstrauch conocido como síndrome neonatal progeroide, en el cual el inicio del envejecimiento comienza en el útero, y los signos y síntomas son evidentes al nacer.
Debe además diferenciarse del síndrome de Werner que comienza en la adolescencia o edad adulta temprana.

Es considerado como síndrome segmentario del envejecimiento, pues pese a que sus síntomas son característicos del envejecer, no presentan un declive de la capacidad cognitiva o incremento de la incidencia del cáncer.